For the impatient
A typical Minia command line looks like:
./minia -in reads.fa -kmer-size 31 -abundance-min 3 -out output_prefix
Type
./miniafor a quick explanation of the parameters.
For more information, refer to the manual.
KmerGenie can be used to determine the best k-mer size, minimum abundance of correct k-mers, and genome size estimation for your dataset.
Support
Please use Biostars (dynamic FAQ system, click "New Post", top right corner) to report bugs or ask any question. (An archive of posts between 2012 and 2014 can be found here.)
Articles
Minia is mainly described in the WABI 2012 article. Different authors proposed and implemented improvements in a WABI 2013 article. Minia now uses these improvements by default.
Original article: Space-efficient and exact de Bruijn graph representation based on a Bloom filter
Abstract
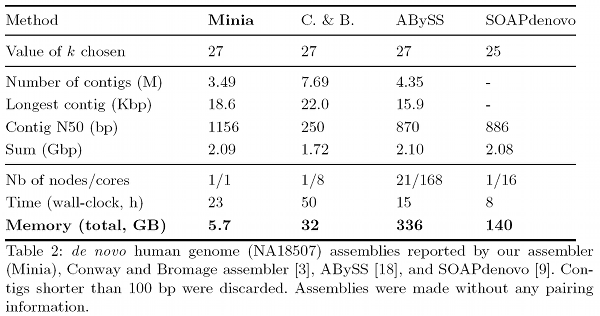
The de Bruijn graph data structure is widely used in next-generation sequencing (NGS). Many programs, e.g. de novo assemblers, rely on in-memory representation of this graph. However, current techniques for representing the de Bruijn graph of a human genome require a large amount of memory (> 30 GB).
We propose a new encoding of the de Bruijn graph, which occupies an order of magnitude less space than current representations. The encoding is based on a Bloom filter, with an additional structure to remove critical false positives. An assembly software implementing this structure, Minia, performed a complete de novo assembly of human genome short reads using 5.7 Gb of memory in 23 hours.
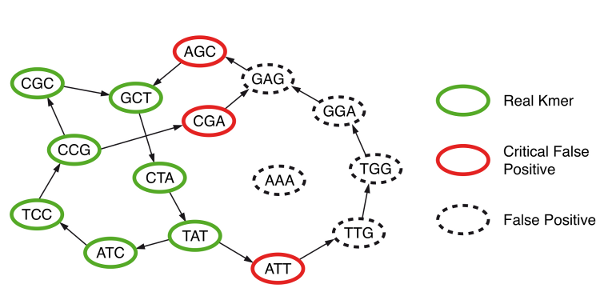
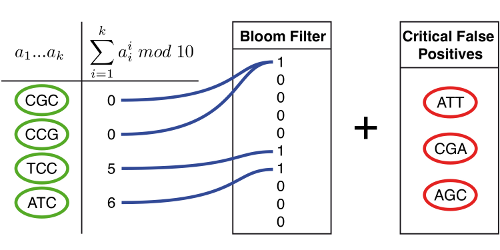
Example
In this example, the de Bruijn graph consists of seven nodes (green). They are hashed and inserted in a Bloom filter. The Bloom filter creates false positives (black and red). Only the false positives that are immediate neighbors of the true nodes are critical for assembly (red). By storing only the Bloom filter and the set of critical false positives, we can perform assembly on the exact de Bruijn graph.
PDF and Citation
R. Chikhi, G. Rizk. Space-efficient and exact de Bruijn graph representation based on a Bloom filter, WABI 2012 (conference version)
@inproceedings{minia,
author = {Chikhi, Rayan and Rizk, Guillaume},
title = {Space-Efficient and Exact de Bruijn Graph Representation Based on a Bloom Filter.},
booktitle = {WABI},
pages = {236-248},
publisher = {Springer},
series = {Lecture Notes in Computer Science},
volume = 7534,
year = 2012
}
Steps to reproduce the human genome assembly from the article
Download these raw human genome reads: SRX016231, convert them to FASTA and concatenate them into a single file (HG_reads_all_untrimed.fa).
Launch Minia as follows:
./minia HG_reads_all_untrimed.fa 31 3 2700000000 results_prefix
Using cascading Bloom filters to improve the memory usage for de Brujin graphs
Abstract
De Brujin graphs are widely used in bioinformatics for processing next-generation sequencing (NGS) data. Due to the very large size of NGS datasets, it is essential to represent de Bruijn graphs compactly, and several approaches to this problem have been proposed recently. In this work, we show how to reduce the memory required by the algorithm of Chikhi and Rizk (WABI, 2012) that represents de Brujin graphs using Bloom filters. Our method requires 30% to 40% less memory with respect to their method, with insignificant impact to construction time. At the same time, our experiments showed a better query time compared to their method. This is, to our knowledge, the best practical representation for de Bruijn graphs.
PDF and Citation
K. Salikhov, G. Sacomoto and G. Kucherov. Using cascading Bloom filters to improve the memory usage for de Brujin graphs, WABI 2013
@incollection{cascading,
title={Using cascading Bloom filters to improve the memory usage for de Brujin graphs},
author={Salikhov, Kamil and Sacomoto, Gustavo and Kucherov, Gregory},
booktitle={Algorithms in Bioinformatics},
pages={364--376},
year={2013},
publisher={Springer}
}